
AS/NZS 3551 and AS/NZS 2500 - ongoing issues
The standards AS/NZS 3551:2012 Management programs for medical equipment and AS/NZS 2500:2004 Guide to the safe use of electricity in patient care purportedly represent the views of clinical engineering consultants and employees of healthcare organisations.
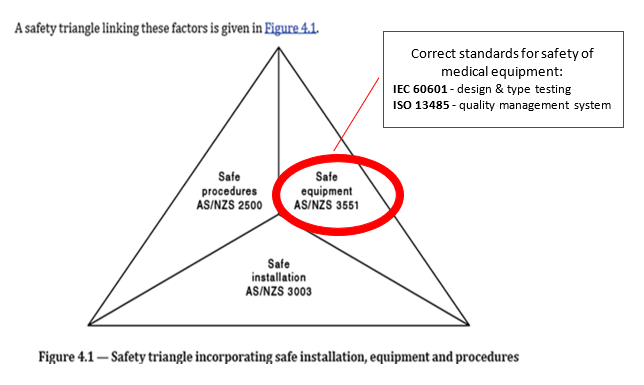
The AS/NZS 2500 includes a so-called “safety triangle” (see Fig. 4.1 reproduced below from the draft document open for public comment) which references AS/NZS 3551 as “the” safety standard for medical equipment. In fact, medical devices must be designed and tested in accordance with type testing standards such as the IEC 60601 family and be developed and manufactured within a quality management system in accordance to ISO 13485. AS/NZS 3551 is only for in-house management of medical equipment with some in-field testing and as such it is aimed at healthcare organisations only.
Because of the incorrect reference, AS/NZS 3551 standard has increasingly been included in invitations to tender by State health procurement organisations as a requirement for suppliers of medical devices to comply with and/or to provide certification of medical devices to this standard.
The applicable standards for medical devices, manufacturers and distributors are:
- Type testing standards such as those in the IEC 60601-1 family of standards
- ISO 13485 for quality management systems of manufacturers
- ISO 9001 for quality management systems of distributors
MTAA has been raising concerns about AS/NZS 3551 since 2015, and in December 2018 submitted a project proposal with the HE-003 technical committee to address misleading or false statements, poor structure and a general lack of clarity and specificity in the requirements of AS/NZS 3551. The MTAA project proposal enjoyed strong and wide support from our membership.
Some of the main problems with AS/NZS 3551:2012 are:
- Section 9 allows modifications (other than custom-made devices)/ refurbishments to be done by hospitals without any safeguards such as ensuring that the basic safety and essential performance of medical devices are still maintained. It states: “If the safety function or operability of the medical device has been changed, a legible and indelible label shall be permanently secured in a place clearly visible to the user.” Clearly, labelling alone is not enough!
- Section 10 fails to specify that if hospitals fail to maintain/ service medical equipment in accordance with manufacturer’s instructions such deviations should be documented and justified.
International best practice requires that healthcare organisations that modify/ refurbish medical devices not on behalf of the original manufacturer, ensure that the medical devices they manufacture/ modify are safe and effective. By undertaking a manufacturer’s activities, they assume the responsibility and liability of a medical device manufacturer.
The new EU Medical Device Regulations (MDR) require health institutions engaging in manufacturing or modifying medical devices for their internal use to meet certain conditions:
- Demonstrate compliance with applicable general safety and performance requirements (the equivalent to Australian Essential Principles)
- Establish an appropriate quality management system (such as those defined in ISO 13485)
- Review experience gained from clinical use of the devices and take all necessary corrective actions
- Justify that the target group’s specific needs cannot be met by an equivalent device on the market
- Make information available to the regulatory authorities upon request
- Make a declaration containing relevant details about the device publicly available
For more details please refer to the MTAA spreadsheet detailing issues with AS/NZS 2500 and 3551 standards.
Australian medical regulations are in the process of being aligned with the EU MDR and it is only a matter of time until this regulatory loophole is closed in Australia as well. Until then, MTAA is calling on all stakeholders – industry, consumer representatives, healthcare providers – to join us and address the current problems in AS/NZS 3551 and AS/NZS 2500 in a constructive and collaborative manner.
References:
In-house service and refurbishment of medical equipment - understanding regulatory risk, Brandwood CKC 2019
Substantial changes affecting a TGA conformity assessment certificate and Transfers of certificates, TGA 2017
Changes in rules for manufacturing or modifying and use of medical devices including in vitro diagnostic medical devices, NHS 2018
FDARA Section 710 Report on the Quality, Safety, and Effectiveness of Servicing of Medical Devices, FDA 2018
